|
Chromatin modifications, such as post-translational modification of histone proteins and incorporation of histone variants, play an important role in regulating gene expression. Individual histone modifications can regulate gene expression by changing chromatin structure and creating binding sites for effector proteins. More importantly, joint analyses of multiple histone modification maps are starting to reveal combinatorial patterns of histone modifications that are associated with functional DNA elements, providing strong support to the unified ‘histone code’ hypothesis. Due to the lack of computational methods, only a small number of chromatin modification patterns have been associated with well-known functional DNA elements, e.g. promoters and enhancers. To develop novel insights into the histone code, we propose a scalable subspace clustering algorithm, Coherent and Shifted Bicluster Identification (CoSBI), to identify the complete set of combinatorial chromatin modification patterns across the entire genome. Comparison of CoSBI with an existing methods demonstrates that our algorithm can generate biclusters with higher intra-cluster correlation and biological relevance. |
 |
|
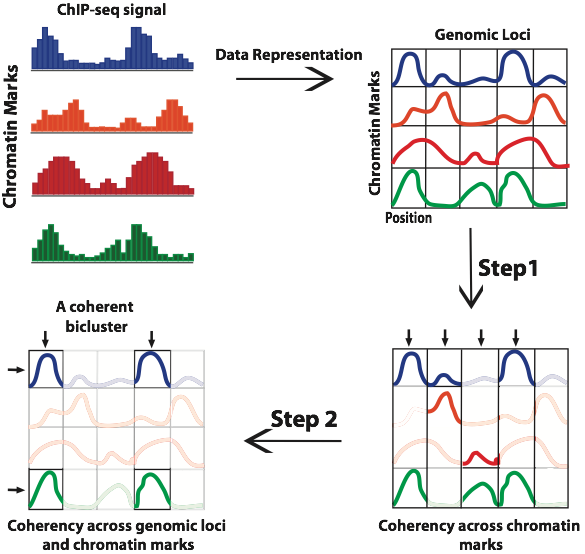
As shown in the above figure we first converts many ChIP-seq/ChIP datasets into a 3D matrix. In the first step of CoSBI, for every genomic locus, it identifies maximal subsets of chromatin modifications that exhibit coherent signals among them. In the second step, the algorithm identifies coherent patterns across both Genomic locus and Chromatin mark dimensions, generating coherent biclusters. The final output of our algorithm is a complete collection of biclusters across the genome, each of which contains a set of chromatin modifications that exhibit coherent signals across all genomic loci in the given bicluster. |
|
You can download the CoSBI package using the link below. This package includes a command-line version of CoSBI (implemented in C++), a user friendly version of CoSBI (implemented in C++ using the Qt package) with a GUI, explanation of the CoSBI algorithm, and example datasets. |
| We applied our algorithm to a compendium of 39 genome-wide chromatin modification maps in human CD4+ T cells. We identified 843 combinatorial patterns that are repeated across at least 0.1% of the genome. You can download these biclusters along with their functional enrichment pvalues as a supplemental table from our NAR paper website. A total of 19 chromatin modifications are observed in the combinatorial patterns, 10 of which occur in more than half of the patterns. Our analysis further reveals combinatorial chromatin modification signatures for 8 classes of functional DNA elements. Application of CoSBI to epigenome maps of different cells and developmental stages will aid in understanding how chromatin structure helps regulate gene expression. |
|
|
|
The CoSBI method was originally introduced and applied to T cell in the following paper: Ucar D, Hu Q, and Tan K. 2011. Combinatorial chromatin modification patterns in the human genome revealed by subspace clustering. Nucleic Acids Res. Email any questions, comments, or bugs found to Kai Tan (tank1@email.chop.edu) |