|
Combinatorial interactions among transcription factors (TFs) are critical for integrating diverse intrinsic and extrinsic signals, fine-tuning regulatory output, and increasing the robustness and plasticity of the regulatory mechanism. Current knowledge about combinatorial regulation is rather limited, due to the lack of suitable experimental technologies and bioinformatic analysis tools. The rapid accumulation of ChIP-Seq data has provided genome-wide occupancy maps for a large number of TFs as well as chromatin modification marks for identifying enhancers without knowing individual TF binding sites beforehand. Integration of the two data types has not been researched extensively, resulting in under-utilized data and missed opportunities. We develop a novel method for discovering frequent combinatorial occupancy patterns by multiple TFs at enhancers. Our method is very useful for studying combinatorial gene regulation taking advantage of increasingly abundant ChIP-Seq data. |
 |
|
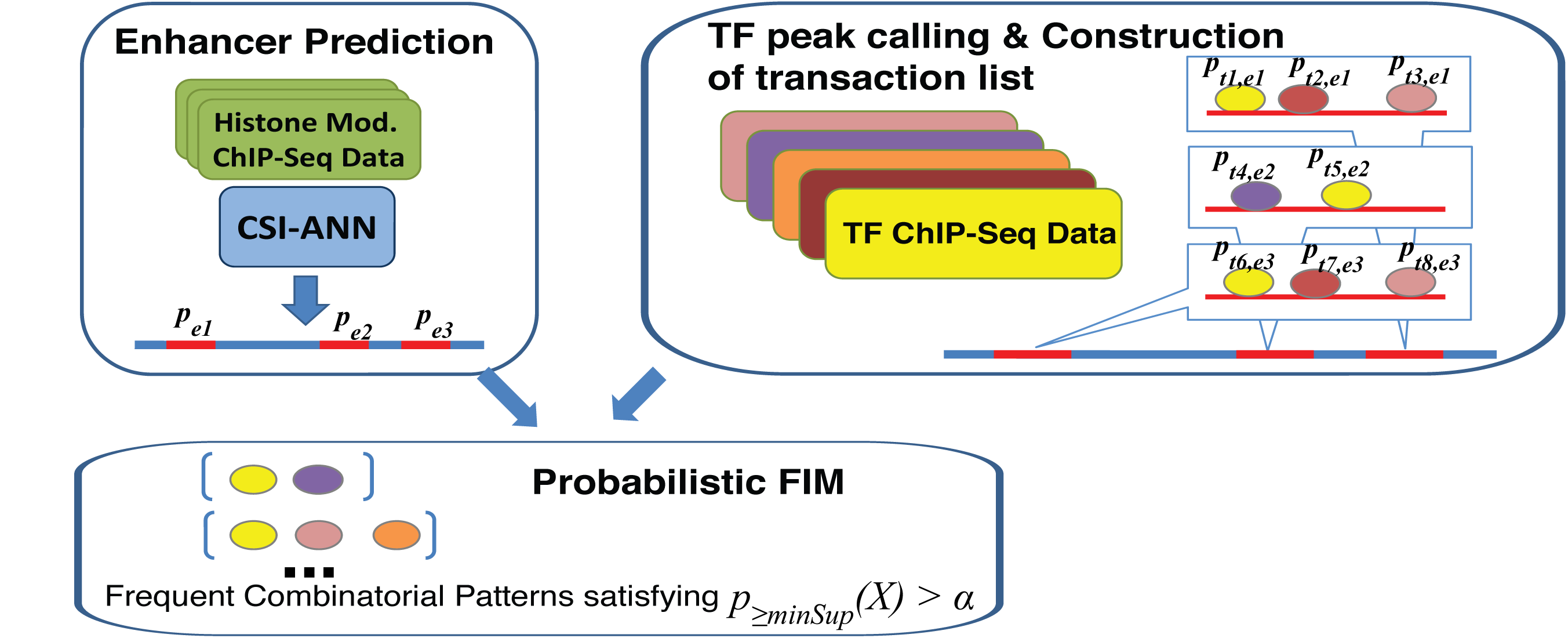
Description of the Algorithm: Our method is motivated by the concept of frequent item set mining (FIM). However, unlike traditional FIM, our method considers the probabilities of both transactions and items, which is a way to deal with uncertainty in ChIP-Seq data. Our method has the following three components: 1) calculation of probabilities associated with enhancers (transactions) and TF binding peaks (items); 2) automatic determination of pattern-specific minimum support threshold; 3) calculation of frequentness probability of candidate FCOPs using an uncertain transaction database and dynamic programming. |
|
Input Data: The input data consists of genome-wide location information for enhancers and multiple TFs and probability values that are associated with each type of sequence. Enhancers can be defined based on co-factor (e.g. P300) ChIP-Seq or based on chromatin modification signature. In our study, we define genome-wide enhancers using CSI-ANN and histone modification ChIP-Seq data. For each predicted enhancer CSI-ANN assigns a probability. The higher the probability the more confidence we have in the prediction. The other required data type is TF Chip-Seq data. Binding peaks for TF can be called using a number of peak callers such as MACS. |
|
The software package can be downloaded using the link below. This package includes a command-line version of the software (implemented in C++), readme file explaining how to run the software, and example datasets. |
|
|
|
The method was originally described in the following paper: L. Teng, B. He, P. Gao, L. Gao and K. Tan 2014. Discovering context-specific combinatorial transcriptional factor interactions by integrating diverse ChIP-Seq datasets. Nucleic Acids Res. 42(4):e24, 2014 Email any question, comment, or bug report to Kai Tan (tank1@email.chop.edu). Go Back to Tan Lab. |