|
In eukaryotes, gene expression is controlled by short regulatory DNA sequences called enhancers. How does an enhancer select its target promoter(s) is a major challenge in the field of gene regulation. Advances in genomic technologies have enabled rapid and comprehensive identification of active promoters and enhancers for many cell types. But there is a lack of methods to link bona fide enhancers and their target promoters. Here, we develop and integrate multiple genomic features into a statistical predictor for enhancer-promoter interactions. Our approach presents a systematic and effective strategy to decipher the mechanisms underlying enhancer and promoter communication. |
|
|
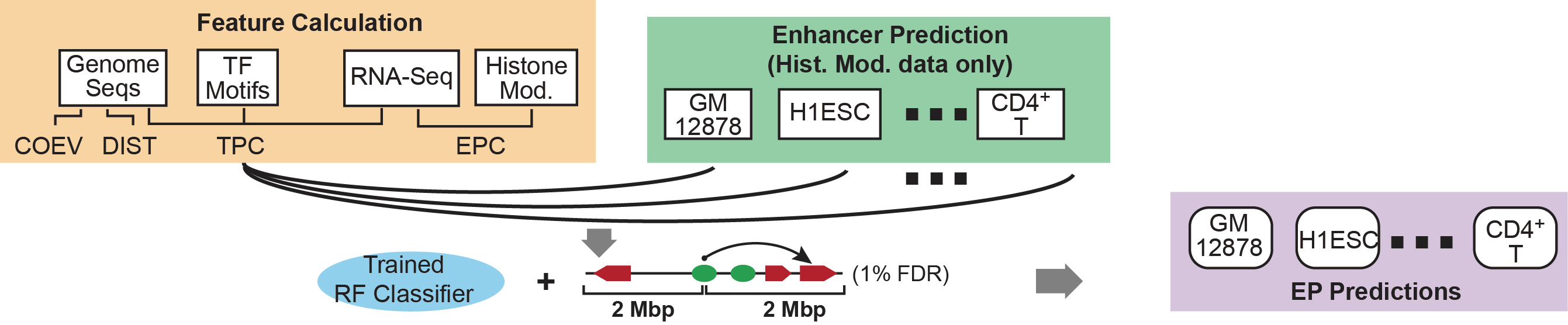
Description of the Algorithm: We devised and tested four features for their abilities to discriminate a set of ~2000 real and non-interacting EP pairs that are selected based on published ChIA-PET data. The four features are 1) distance constraint (DIS); 2) enhancer and target promtoer activity profile correlation (EPC); 3) TF and target promoter correlation (TPC); and 4) co-evolution of enhancer and target promoter (COEV). All these genomic features can be extracted from public database. For genome-wide prediction of EP pairs, we first use CSI-ANN and histone modification ChIP-Seq data (specific to each cell type) to predict enhancers. For each predicted enhancer we extract all candidate promoters within a distance range (e.g. 2M bp) and compute the four feature scores separately for all EP pairs. We next use random forest classifier to integrate the four features for linking enhancers to their target promoters. |
|
Input Data: There are three required input files for genome-wide prediction of EP pairs by IM-PET: 1) enhancer positions; 2) genome-wide enhancer signals; and 3) gene expression information. The first 2 files are the output directly from CSI-ANN, which uses histone modification ChIP-Seq data to predict enhancers. We recommend using the combination of H3K4me1, H3K4me3, and H3K27ac as it mark active enhancers. Users can also use fewer number of histone marks (e.g. combination of H3K4me1 and H3K4me3) or even DHS sites. For gene expression information, we recommend RNA-Seq data as this type of data can distinguish different transcripts better than microarray data. However, microarray data is also applicable. We recommend the user to process the RNA-Seq data using Tophat and Cufflinks software. IM-PET uses as input one of the output files of Cufflinks "isoforms.fpkm_tracking". |
|
Access to the Software: |
|
|
|
|
|
|
Email any question, comment, or bug report to Kai Tan (tank1@email.chop.edu). Go Back to Tan Lab. |